Our four Specific Aims
Aim 1
Enabling methods for highly multiplexed changes to genomes to test causal hypotheses flowing from genome sequencing and human traits (especially cis-regulatory variation effects on gene expression).
Aim 2
Automating establishment of human induced Pluripotent Stem cells (iPS), differentiated cell types and complex in vitro tissues; and engineering heterozygosity in multiple exons of genes to enable analysis of allele-specific transcription and splicing.
Aim 3
Exploring bar-coding, in situ RNA multiplexing, and single-molecule approaches to monitor the impact of genetic variations in many RNAs and many cell types at once.
Aim 4
Developing methods for low-cost synthesis of long DNA constructs, efficient homologous recombination in human cells, and highly multiplexed single cell handling that enables sorting based on morphology.
Aim 1
We will develop and demonstrate novel methods that identify and characterize natural cis variations that directly affect transcriptional activity in individual humans based on direct modification and testing of combinations of variants in gene regulatory regions in cell lines, and that can be applied to thousands of genes.
1.1. We will develop and demonstrate novel, high-efficiency methods to create human cell populations containing combinations of natural variations in gene regulatory regions, focusing on zinc-finger nuclease (ZFN)-mediated recombination of externally generated altered insert libraries, and direct modification of human cells using oligo-based methods.
1.2. We will demonstrate the identification of specific sets of variations that affect cis gene transcription by engineering many combinations of variations and directly observing their effects on transcription, and also by novel methods of assaying complex populations of combinatorially modified cells at a single-cell level.
1.3. We will assess the extent to which cis variants identified as causing altered transcript expression may operate through alternative mechanisms such as differential expression of RNA isoforms, differential transcript degradation, copy number variations, and epistatic marks.
1.4. We will analyze the relationship between our methods and results and those of Genome Wide Association Studies and characterize their complementary insights into the effects of variation.
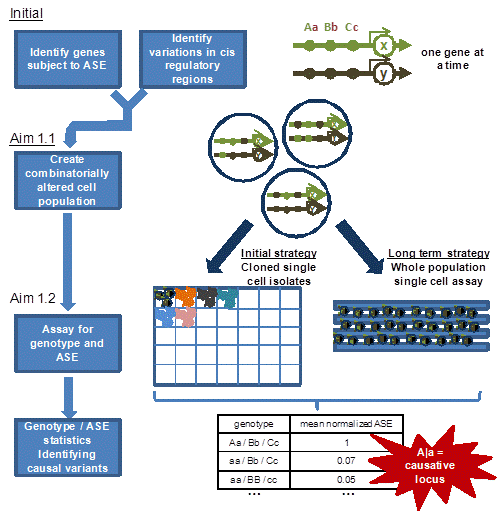
Overview of Aim 1 strategy for identifying causative cis variations. Initial Aim 1 work identifies genes subject to allele-specific expression (ASE), and, via next-gen sequence data, also identifies variations in regulatory regions, e.g., here the 100kpbs upstream region. Via Aim 1.1, cell populations are created for each gene bearing combinations of the variations identified for that gene. Via Aim 1.2, cells from this population are genotyped and assessed for ASE so that the specific loci and loci interactions that control ASE can be identified. The initial Aim 1.2 strategy will examine clonal outgrowths of individual altered cells from the population, while a longer term strategy will assay the entire mixed altered population at a single cell level. This strategy is executed one gene at a time for 100s to 1000s of genes.
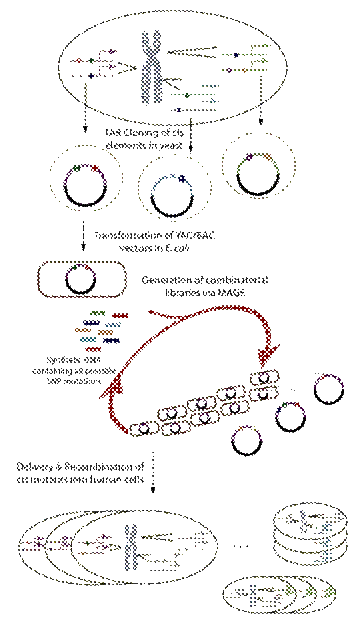
One method that will be developed to create libraries of altered cis variation loci in regulatory regions of a gene. Large regulatory fragments will be moved to yeast via TAR cloning and from there to E. coli as BACs. Multiplex Automated Gene Engineering (MAGE) will be applied to create alterations in the targeted cis variations on the BACs, after which parts or wholes of the altered regions, or entire combinatorial libraries, will be transferred back to the original human cells to form cis-altered populations of cells.
Aim 2
We will adapt and extend Aim 1 methods to function in human induced Pluripotent Stem cells (iPS) and then use iPS to characterize the effect of cis regulatory region variations in a variety of derived cell types that represent different human tissues. We will engineer “marked allele” human iPS that are heterozygous in all exons of many genes that will enable analysis of allele-specific transcriptional and splicing effects in diverse cell types.
2.1. We will combine Aim 1 methods with automated techniques for iPS generation and maintenance to enable exploration of iPS with altered cis regulatory regions.
2.2. We will differentiate iPS generated in Aim 2.1 into diverse cell types that represent distinct human tissues and characterize the cell type-specific consequences of cis-regulatory variations.
2.3. We will engineer human iPS with “marked alleles” for 10-50 genes and demonstrate their use by characterizing allele-specific transcription and splicing in multiple tissues.
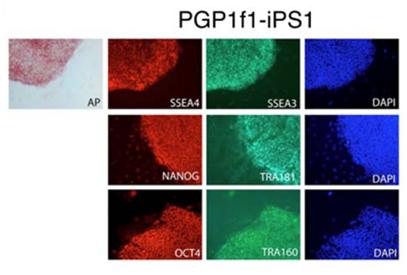
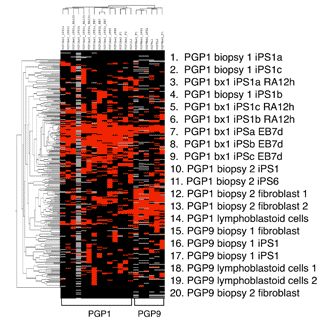
Allele-specific expression (ASE) in induced Pluripotent Stem cells (iPS) and derivatives developed from a Personal Genome Project (PGP) participant. Left: iPS cells expressed molecular pluripotency markers (SSEA4, SSEA3, Tra1-60, Tra1-81, NANOG and OCT4) and stained for alkaline phosphatase activity. When injected into immune-deficient mice, iPS cells formed a teratoma, containing normal tissues from all three germ layers, including respiratory epithelium (endoderm), bone (mesoderm) and neuroectoderm (ectoderm). (See article). Right: Hierarchical clustering of statistically significant (ASE) in reprogrammed cells from two PGP subjects, showing consistent but genotype-specific ASE signatures across different cell types, culture conditions, and cell batches.
Aim 3
We will develop novel single-cell in-depth transcriptome assays scalable to millions of individual cells simultaneously in both structured tissues and dispersed cell samples, subject to sequencing capacity. These methods will be used to explore systematic transcriptional effects of genetic variations in different human cell types.
3.1. We will develop and optimize methods that pipeline in-situ single-cell cDNA synthesis to next generation sequencing in ways that preserve cell identity and that can be applied in parallel to 100s to 1000s of cells. We will investigate multiple techniques in support of these methods, including cell bar-coding, in-situ cell sequencing, and single-molecule in-cell sequencing, characterize their performance and limits, and select one for continued development and application.
3.2. We will use these single cell transcriptomics capabilities to characterize the transcriptional state differences in cells bearing artificial and natural variant combinations from Aim 1, and from cell types developed from iPS from different genetic backgrounds.
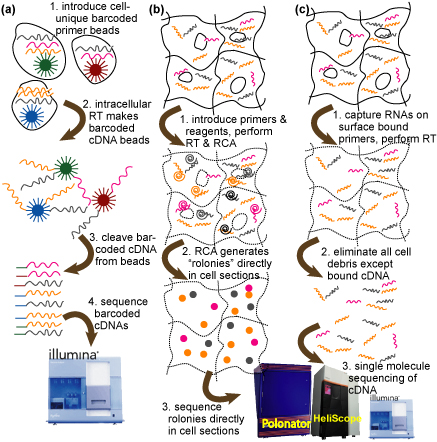
Overview of single cell transcriptomics approaches explored in Aim 3. (a) Barcoding: RT primers and unique sequence barcodes are created on microbeads that capture mRNAs from single cells. cDNA is synthesized on the beads, and the beads are extracted. The barcoded cDNAs are cleaved off the beads and sequenced directly on a next generation sequencer. (b) In-situ RCA sequencing: Reagents and primers are introduced into permeabilized cell sections in order to perform RT and rolling circle amplification of cDNA fragments, forming ‘RCA colonies’ or “rolonies”. Cell debris is removed and rolonies are directly sequenced in cell sections. (c) Single molecule: As in (b) cell sections are permeabilized, but first strand cDNA is created directly on surface-anchored primers and sequenced directly without amplification using single molecule techniques.
Sequencing capacity considerations: Given 100Gbp run (2009 Illumina goal), approach (a) could yield ~1e4 single cell transcriptomes or ~1.8e5 single cell sub-transcriptomes covering 1000 specific genes. See Figure 5.3-1 in our proposal for details.
Sequencing: (a) can be performed on any current next gen sequencer, but (b) and (c) will require modified technology. We will develop this technology on the Polonator and migrate to other instrumentation.
Aim 4
In support of Aims 1-3, we will develop innovative and widely applicable methods for high-throughput synthesis of long DNA constructs, highly efficient homologous recombination in human cells, and highly multiplexed single cell handling that enables sorting based on morphology.
4.1. We will develop a platform that integrates DNA synthesis and sequencing and uses sequence information to assure synthesis of DNA constructs with extremely low error rates.
4.2. We will improve ZFN–mediated homologous recombination in human cells by engineering a comprehensive zinc-finger archive, by developing novel methods of delivering ZFNs into cells, and by developing a “segmental genome replacement” strategy.
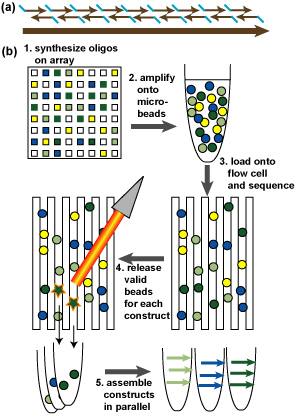
One method we will develop in Aim 4,1 for integrating DNA sequencing and synthesis and enabling high-throughput reduced-error synthesis of large constructs. (a) Large DNA construct is analyzed into oligos with appropriate overlaps, uniqueness, Tms, as needed. (b) Processing pathway from synthesis of oligos on array for multiple constructs (represented by different colors) to multiplex synthesis. Amplfication in (2) is illustrated as emulsion PCR as in (reference). Microbeads are loaded onto flow cell using light-labile chemical attachments and sequenced on the flow cell (3). For each construct, light is directed to microbeads with sequence-validated oligos for the construct for release and capture (4). Assembly of all constructs then proceeds in parallel (5).